Ionization Mechanisms in UV-MALDI
Previous: Rate Equations Table of Contents Next: Surfaces
5. The Quantitative CPCD Molecular Dynamics Model
Motivation for MD
Implementation
Ion Recombination
Results
Movies
Why also Molecular Dynamics?
The rate equation implementation of the CPCD has been remarkably successful, considering its simplicity. Although clearly useful based on its good correlation with experiment, it required some simplications with respect to the desorption/ablation event. It assumes that a sample layer completely changes phase when it reaches a specified temperature. From that point it is treated as an expanding gas. The ion yield is then reduced by a factor compensating for the expected fraction of condensed material (clusters, droplets, chunks).
Both the phase change temperature and the cluster fraction can be estimated from experimental data, but these approximations are rather crude compared to the true vaporization/ablation/spallation nature of the early plume. It is important to better understand the interaction of ionization processes in this complicated early period of MALDI.
Molecular level models of MALDI vaporization were investigated in 1998-2004, but these did not include ionization processes. A combined model was therefore developed in 2005. See
J. Phys. Chem. B, vol. 109, pp. 22947-22957 (2005)
(final submitted
manuscript)
and
J. Mass Spectrom., vol. 45, pp. 333-346 (2010)
(final submitted
manuscript,
another source)
Implementation
The mechanical aspects of this molecular dynamics (MD) model
had been developed in detail by Zhigilei and Garrison. Their model had
the special advantage that the smallest particle is a molecule, not an
atom. They realized that the details of molecular vibrations in the
thick MALDI plume were not very important. It is only necessary to
include one effective internal degree of molecular freedom, so that
translational and vibrational energy can interconvert in collisions.
This simplification has the major advantage that the time step and the
physical dimensions of the simulations can be orders of magnitude
larger than for traditional MD. See, for example, Zhigilei,
Leveugle, Garrison, Yingling, Zeifman, Chem. Rev. vol. 103, p. 321
(2003).
While the ionization processes in the MD model are exactly the same as in the rate equation model, they had to be converted to a probability form, rather than a rate form. In other words, the intermolecular interactions between matrix and analyte molecules must be specified, in detail, for the various states involved. This makes the model somewhat less general than the rate equations. For example, pooling of excited states are taken to be due to the interaction of matrix molecular dipoles, similar to Förster energy transfer. The potential for this is proportional to 1/R6, where R is the intermolecular distance.
Parameters were sometimes available directly from experiment, others were derived indirectly from those determined for the rate equations. Some were new to this model. The list below describes some of the necessary values. At the moment, the parameters have been determined only for DHB matrix, but many others are expected to be similar.
The simulations performed to date typically include a 10 X 10 X 360 (depth) nm block of material, periodic boundary conditions are applied in the lateral directions. Underneath the material, a continuous, elastic material is simulated, allowing for propagation of pressure pulses out of the material and for rebound afterward. The simulation time step is a few femtoseconds, and simulations of several to tens of nanoseconds are feasible, with computation times of about weeks to months on desktop computers.
Ion-Ion Recombination by Tunneling
As noted in the discussion of the rate equation model, loss mechanisms are a very
important part of the MALDI process, especially for determining the final ion yield.
MD is a good way to investigate this key process, as reported in
Analytical and Bioanalytical Chemistry, vol. 402, pp. 2511-2519 (2012)
(final submitted manuscript).
In the MD model, it is necessary to specify the probability of recombination as a function of distance between two oppositely charged ions. Initially a Langevin model was used, as is often applied in solid-state physics. In the Langevin model, the recombination rate is assumed to be dominated by the rate at which the ions find each other. In this case one can assume that when they reach some reasonable distance they "touch" and charge is transferred. The key parameter is this threshold distance, which is "hard", or infinitely sharp.
As the MD simulations quickly show, the charge density in the sample before ablation can be quite high. Ions are already in close proximity, the rate at which they find each other is not limiting. A first step away from the hard edge model might be a soft edge. This was tested and found to be not much better than the hard edge.
Some reflection on the approach of two ions leads to a different, more physically based model: As the ions approach, how will charge finally be transferred? If the ions are radicals, it is obviously an electron that will jump. If they are proton or other adducts will it be the adduct ion? No, because the electronic wavefunctions of the molecules will begin to interact long before the atoms are "ready" to move. To put it another way, the electron has a vastly larger quantum mechanical tunneling range than does an atom.
So the far more probable process is electron transfer followed by neutral atom rearrangement. The ions are first neutralized by electron hopping, then the atoms move (comparatively) slowly to adapt to the new charge distribution:
MH+ + (M-H)- —> MH° + (M-H)° —> M + M
Electron tunneling has an exponential dependence on intermoleculear distance, as well as a dependence on the intramolecular rearrangements that take place when an electron is added or taken away. Fortunately, there is a huge literature on electron transfer processes, and some very widely applicable generalizations have been derived. These were incorporated into the model, and found to give very reasonable results:
log10(tunneling rate) = α - βR - K(ΔG + λ)2/λ
R is the intermolecular distance. The α parameter corresponds to a typical frequency factor in reaction rate theory, and is typically 12-13. The decay exponent β, depends on the height of the barrier, and is typically 0.5 - 1.1. The reorganizational energy, λ, depends on the specific nature of the donor and acceptor molecules and their vibronic potential surfaces. since this term is not dependent on intermolecular distance it has the same effect as reducing α.
Just as important, variation of the tunneling parameters was found to cause a significant change in the rate of initial ablation. This suggests that photoacoustic pump-probe techniques could be used to investigate and possibly validate or characterize this aspect of MALDI.
Finally, if this concept is correct, one of the properties of matrix molecules
that is relevant for their MALDI performance is how they engage in electron tunneling
with analyte and matrix ions.
Results
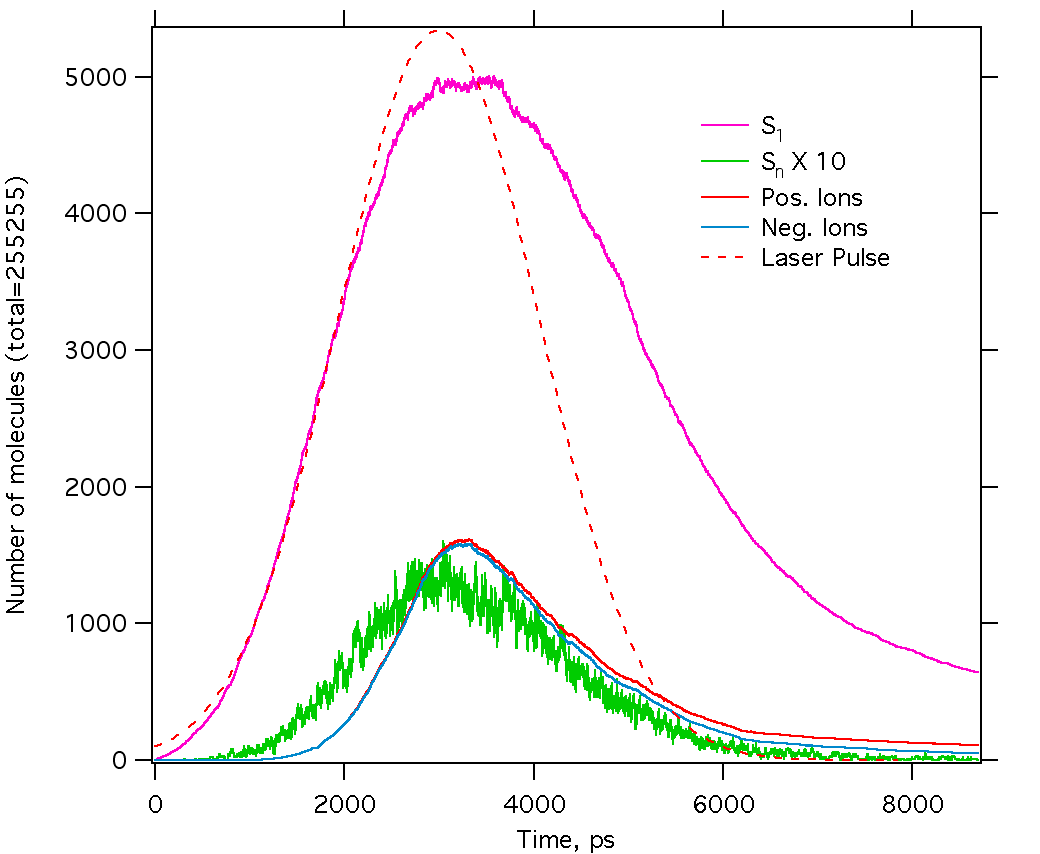
As seen in the next Figure, the evolution of the excited states is
broadly similar to that of the rate equation model (Fig 11). The largest difference
is that the ion populations vary smoothly, without the restrictions
imposed by the assumptions regarding the phase change. The general
features are the same, however. The ion population increases
dramatically during the high density, high excitation period, then
decays as recombination occurs in the expanding material. The curves
are much "noisier" than in the rate model, since only 255255 molecules
are simulated, not a large ensemble. The difference between positive
and negative ions is due to electron escape from the top (and a few
from the bottom) of the material.
Figure 16. Calculated state populations vs. time for MALDI molecular dynamics model. The 355 nm laser pulse had a width of about 3 ns and a fluence of 40 mJ/cm2. There were 255255 molecules in the simulation.
Near the peak of the ion population, at 3 ns, Most of the laser energy is still stored in the electronic excited states, so the material has not heated or expanded much. The ion density is largest near the surface, where the laser intensity is highest. A positive ion has desorbed from the surface, as have several neutral molecules:
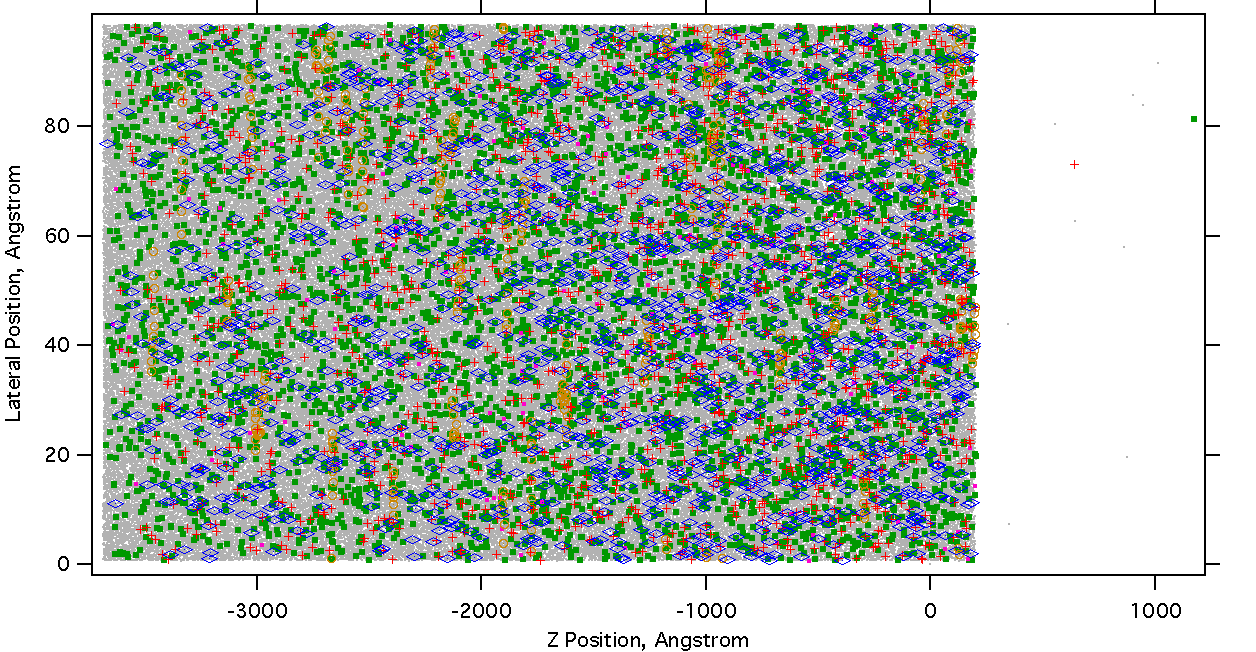
Figure 17. Snapshot at 3 ns, near the peak of the ion population. The laser pulse was incident from the right. Note that the horizontal and vertical scales are very different. Gray dots are ground state (S0) matrix molecules, green are singly excited (S1) and pink are doubly excited (Sn). Positive ions are red crosses, negative ions are blue diamonds. Yellow circles are subunits of analyte molecules, 10-element polymers with properties like a peptide.
Two nanoseconds later, as the laser pulse is waning, much more energy has been converted to heat. The material has expanded considerably, and the top layers are ablating in a phase explosion. Most ions have recombined in the liquid/gas regions, only the solid regions still support high ion density. The first ions ejected are positive, from the charged surface. The surface becomes positively charged because electrons can escape from the top 10 or so nm.
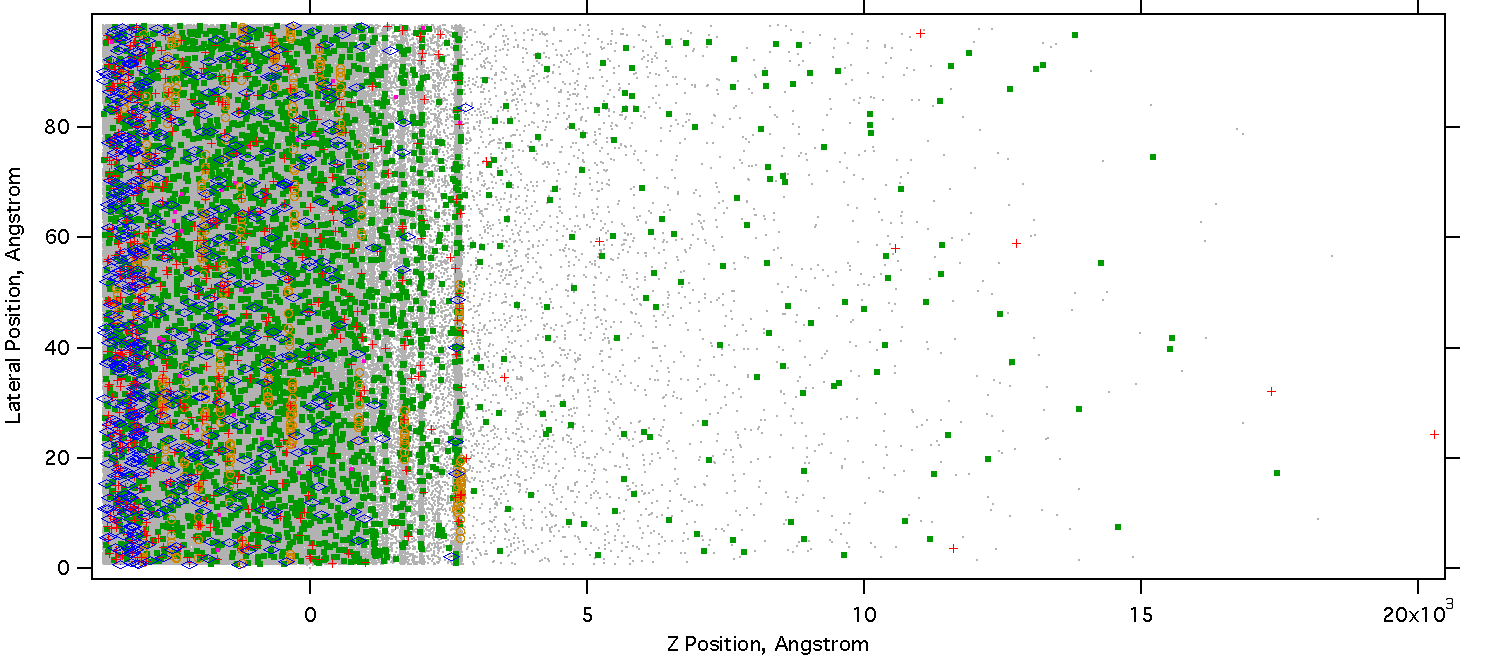
Figure 18. Snapshot at 5 ns, the laser pulse is nearly finished.
At 7 ns, the plume is well developed, and ions are being carried away in both the gaseous and condensed components of the plume. Note the high gas density in the regions between the clusters.
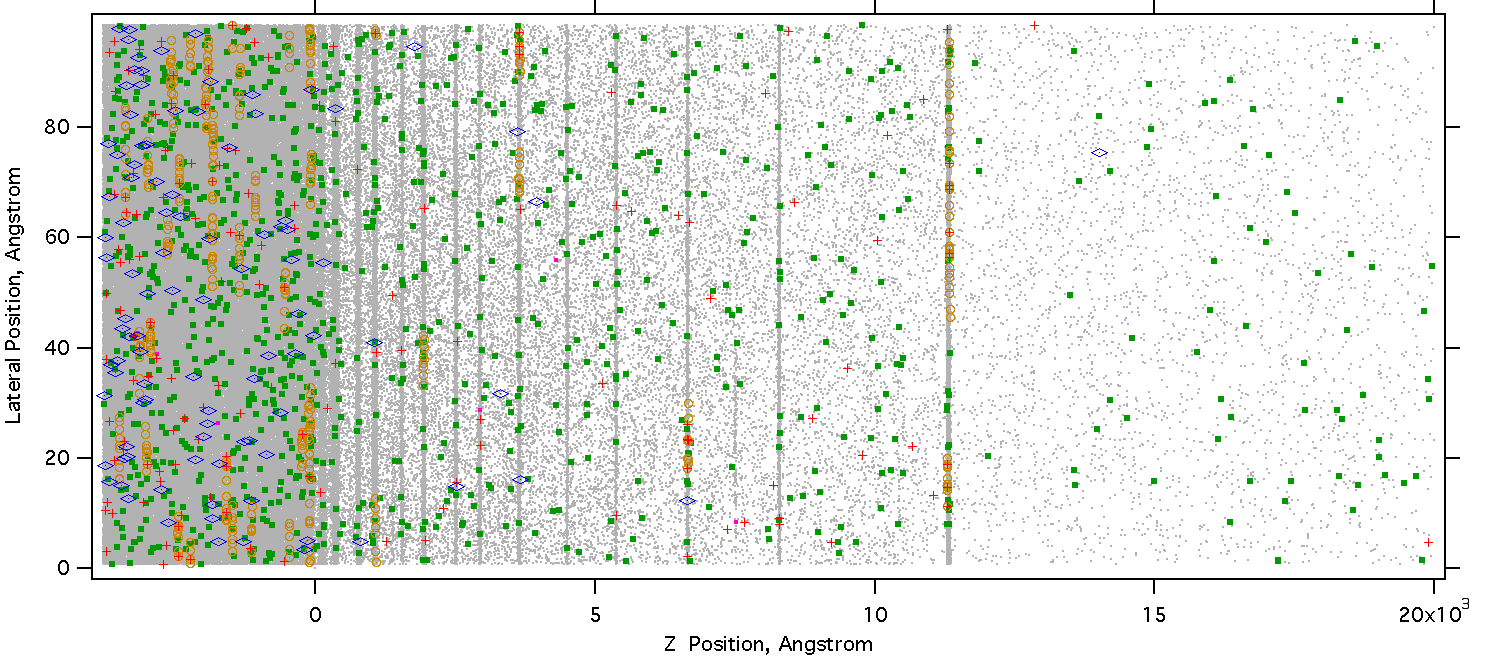
Figure 19. Snapshot at 7 ns, the plume is well developed.
The next figure shows how the clusters lift off the surface, the top layers are accelerated to higher velocities than the lower ones. Note that the clusters are only slightly warmer than the surrounding gas, and that the material cools slowly as it expands. The relatively long, weak laser pulse did not create a sufficiently strong pressure wave to induce spallation in deep, cold layers. Only phase explosion ablation is observed.
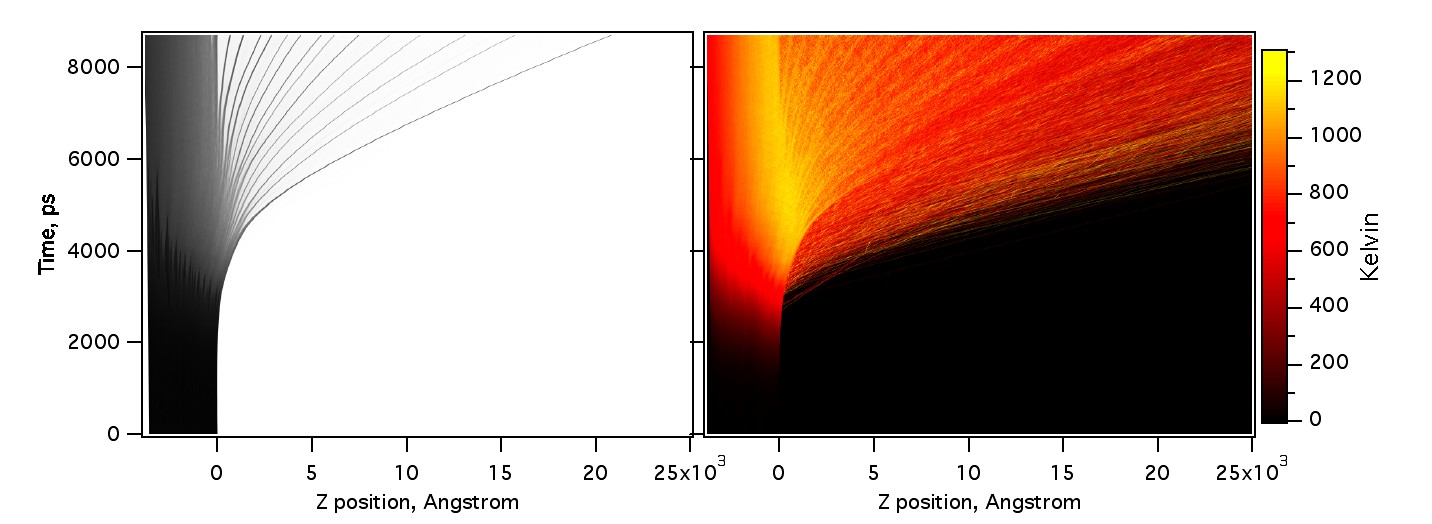
Figure 20. Density (left) and temperature (right) vs time and axial position.
The next Figure shows a zoom of the surface during the development of ablation by phase explosion. The extremely high gas density, with corresponding collision rates, is apparent. The positive surface charging by electron escape is also prominent.
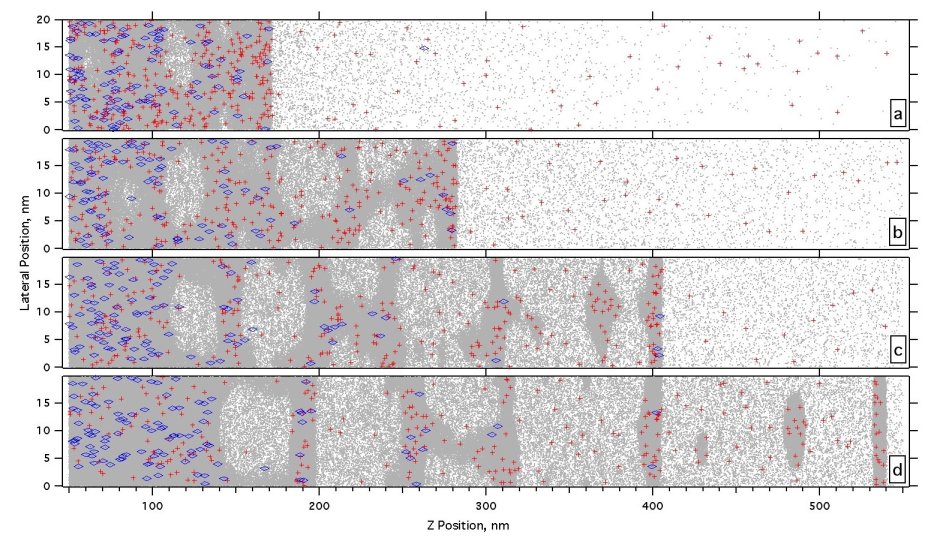
Figure 21. Development of subsurface nucleation in the phase explosion region.The snapshot times are (a-d) 500, 750, 100 and 1250 ps after a 35 ps 355 nm laser pulse.
The MD model has been featured twice on the cover of major journals. See the Literature page for references and links.

Figure 22. Journal covers featuring the MD MALDI ionization model. Left the Journal of Physical Chemistry B, vol. 109, 2005 (final manuscript). Right Journal of Mass Spectrometry, vol. 45, 2010 (final manuscript).
MD Movies
These are large Quicktime files showing the results of MD MALDI
simulations. The laser pulse was in most cases 35 ns, 355 nm. In the upper right
corner, the time is shown in picoseconds from the start of the
simulation. The laser pulse is centered at about t=40 ps. Note that the
axial and lateral scales are very different, for visualization
purposes.
NoMrkSmall.mov 230 MB. Movie showing all molecules as blue dots. This is best for viewing the phyiscal expansion, desorption and ablation process. See the other movies to view the various excited states and ions.
MrkVSmall.mov 94 MB. Smallest movie showing excited states and ions. Gray dots are ground state (S0) matrix molecules, green are singly excited (S1) and pink are doubly excited (Sn). Positive ions are red crosses, negative ions are blue diamonds. Yellow circles are subunits of analyte molecules, 10-unit polymers with properties like a peptide.
MrkSmall.mov
323 MB. Same as
previous, but similar size to movie without excited states.
P & D 48 MB
Laterally averaged pressure and density vs. axial position. Note the
pressure pulse induced first by stress-confined laser heating, then by
ion recombination energy following melting. This is followed by the
tensile stress of relaxation, which induces spallation in the lower
layers. Upper layers depart by subsurface nucleation and ablation.
T & D 65 MB
Laterally averaged temperature and density vs. axial position. Note
that gas and liquid are in good equilibrium, except for the spallation
chunks. However, these also become equilibrated as time progresses.
P & T of gas 231 MB
Laterally averaged pressure and temperature of gaseous material only.
The condensed material is suppressed. The peak gas pressures reach 4-5
MPa, or 40-50 bar.
Charge & D 158 MB Laterally
averaged charge and material densities vs axial position. This is for a
"slow" laser pulse of 3ns duration, centered around 4 ns. The pressure
pulses are therefore smaller, and no spallation is observed. Note the
high charge density before melting, followed by recomination in the
liquid. Charge which reaches the end of the simulation is not
predominantly carried by clusters or condensed particles, but rather is
in the gas phase. Most of the free ions are generated early.
Previous: Rate Equation Model Next: Surfaces
MALDI Ionization Tutorial © Copyright 2007-2016 Richard Knochenmuss